 Von Recklinghausen disease or Neurofibromatosis type 1 (NF1) is a clinically heterogeneous,
neurocutaneous, genetic disorder characterized by café-au-lait spots,
iris Lisch nodules, axillary and inguinal freckling, and multiple
neurofibromas (benign nerve sheath tumors).
Von Recklinghausen disease or Neurofibromatosis type 1 (NF1) is a clinically heterogeneous,
neurocutaneous, genetic disorder characterized by café-au-lait spots,
iris Lisch nodules, axillary and inguinal freckling, and multiple
neurofibromas (benign nerve sheath tumors).Frequency is reported to be 1/3,000 live births. NF1 is reported in many ethnic groups and affects males and females equally.
Neurofibromatosis 1 is an autosomal-dominant disorder (one only need to get the abnormal gene from one parent in order to inherit the disease).
Symptoms are highly variable, even within the same family.
Multiple café-au-lait macules are found in almost all patients (some at birth and most before the first year). Intertriginous freckling develops starting at 5 years. Multiple cutaneous and subcutaneous neurofibromas develop in adults. In older patients, they keep increasing in number and size. Cutaneous neurofibromas do not become malignant. Plexiform neurofibromas (growing along the nerve and its branches) may cause disfigurement, pain, and functional problems and are usually present at birth and may become malignant later in life.
Ocular manifestations include optic pathway gliomas and iris hamartomas (Lisch nodules). Optic pathway gliomas usually develop before age 6 years, and rarely progress thereafter.
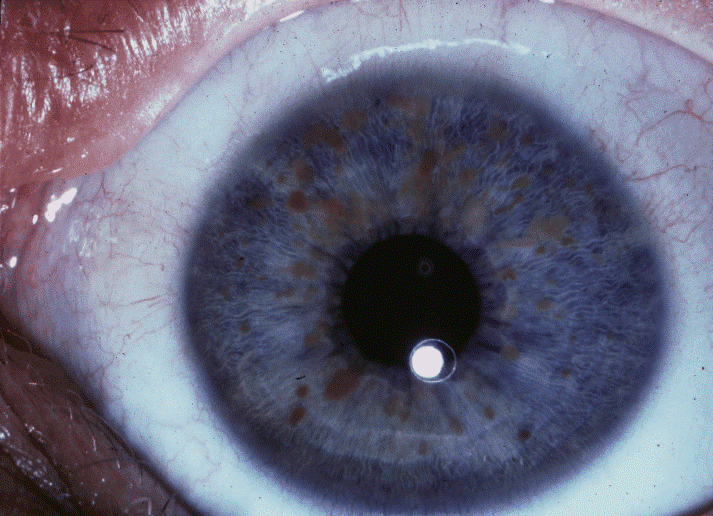 |
| Lish nodules |
Osteopenia (lower bone density), osteoporosis, bone overgrowth, short stature, macrocephaly (big head), scoliosis, skeletal dysplasia (sphenoid wing, vertebral), and pseudoarthrosis (cervical or lumbar fusion in which there is incomplete incorporation or healing of the bone) may be present. Other features include hypertension, vasculopathy, intracranial tumors, malignant peripheral nerve sheath tumor (MPNST; see this term), and occasionally seizures or hydrocephalus. Intellectual development is usually not severely affected but cognitive deficits and learning difficulties are frequent (50%-75%). The overall cancer risk is higher than the general population (lifetime risk of 10-12% for MPNST, mostly between 20-40 years; increased risk of breast cancer before age 50). Three variant forms have been described: familial spinal neurofibromatosis, segmental neurofibromatosis, and 17q11 microdeletion syndrome. Watson syndrome forms part of the NF1 spectrum. Neurofibromatosis-Noonan syndrome is a variant of NF1 in 99% of cases (see these terms).
NF1 is caused by mutations in the tumor suppressor neurofibromin 1 NF1 gene (17q11.2) and rarely by 17q11 microdeletion (only 5%).
Formal diagnostic criteria have been established. 2 or more of the following are diagnostic:
- more than 5 café-au-lait macules,
- 2 or more neurofibromas
- one plexiform neurofibroma,
- optic glioma,
- freckling
- 2 or more Lisch nodules,
- specific bone dysplasias,
- first-degree relative.
MRI can determine the extent of plexiform neurofibromas. Molecular genetic testing can be requested but is mostly not needed.
Legius syndrome (SPRED1 gene) is often clinically indistinguishable from NF1 and is seen in about 2% of people fulfilling NF1 diagnostic criteria. There are however a small number of individuals with NF1 who also do not develop other non-pigmentary manifestations, like Legius syndrome. Constitutional mismatch repair deficiency syndrome (CMMR-D) should be considered. Other differential diagnoses include McCune-Albright syndrome, Noonan syndrome with lentigines (LEOPARD syndrome) and Proteus syndromes. Most cases of Jaffé-Campanacci syndrome are cases of NF1 (see these terms).
Prenatal and preimplantation genetic testing for at-risk pregnancies is possible
The mode of inheritance is autosomal dominant. 1 in 2 cases is caused by de novo NF1 mutations. Penetrance is 100% but disease manifestations vary widely, complicating genetic counseling.
Specific cardiovascular, ocular, neurological and orthopedic manifestations should be treated by corresponding specialists. Cutaneous or subcutaneous neurofibromas can be removed surgically. Plexiform neurofibromas are far more difficult to treat.
Overall prognosis is good but significant morbidity is common. MPNST generally has a poor prognosis. Malignancy and vascular disease are the most common causes of early demise.
Factors associated independently with mortality are the presence of subcutaneous neurofibromas, the absence of cutaneous neurofibromas, and facial asymmetry.