
Frequency is estimated at 1/53,000 and annual birth incidence at 1/36,000. Men and women are equally affected. Mean age at diagnosis is 26 years (range: infancy - 7th decade).
Retinal hemangioblastomas (tumors that originate from the vascular system of the retina) are the most common presenting feature (multiple and bilateral in about 50% of cases). They are usually asymptomatic, but they can cause retinal detachment, macular edema, glaucoma, and vision loss. Retinal capillary hemangioblastomas (RCH) (also known as retinal angiomas) are typically a sign of a von Hippel-Lindau (VHL) disease although they may be seen as an isolated entity. It is the most frequent and earliest manifestation of the VHL disease.
Central nervous system (CNS) hemangioblastomas are the presenting feature in about 40% and occur overall in 60-80% of patients. They are most often located in the cerebellum, but also in the brainstem and spinal cord. They are benign but cause symptoms by compressing adjacent nervous tissue. In the cerebellum they are most often associated with increased intracranial pressure, headaches, vomiting, and limb or truncal ataxia.
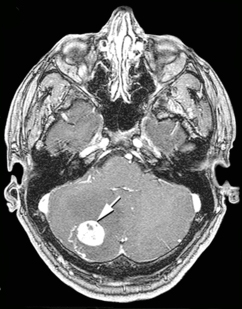 |
| Hemangioblastoma of cerebellum. CT scan. |
Multiple renal cysts are very common and the lifetime risk of renal cell carcinoma (kidney cancer) is very high (70%).
Some patients have pheochromocytomas that can be asymptomatic (no symptoms), but may cause hypertension.
Epididymal cysts and cystadenomas (60% of male patients) may occur, as well as multiple pancreatic cysts (most patients) but non-secretory pancreatic islet cell tumors only occur in a minority (about 10%). Endolymphatic sac tumors (ELST) are also been found (up to 10%) and may cause hearing loss. Head and neck paragangliomas are rare (0.5%).
The mean age at diagnosis of tumors in VHL is considerably younger than in sporadic cases. Marked intrafamilial variability is reported.
VHL is caused by highly penetrant mutations in the VHL gene (3p25.3), a classic tumor suppressor. Most cases are diagnosed via a germline mutation.
Diagnosis can be made in the presence of a single typical tumor (e.g. retinal or CNS hemangioblastomas or RCC) and a positive family history of VHL. If there is no family history (about 20% de novo), multiple tumors (e.g. two hemangioblastomas or a hemangioblastoma and an RCC) are required for diagnosis. A complete blood count, measurement of urinary catecholamine metabolites, urinalysis, and urine cytology may be indicative of polycythemia, pheochromocytoma, renal anomalies, and RCC. Imaging studies can be used to detect CNS tumors, pheochromocytoma, endolymphatic sac tumors, renal tumors, and renal and pancreatic cysts.
Differential diagnoses include multiple endocrine neoplasia, neurofibromatosis, polycystic kidney disease, tuberous sclerosis, Birt-Hogg-Dube syndrome, and hereditary pheochromocytoma-paraganglioma syndromes (see these terms) associated with succinate dehydrogenase subunit mutations (SDHB, SDHC and SDHD).
Antenatal diagnosis is possible through molecular analysis of amniocytes or chorionic villus cells if a disease-causing mutation has been identified in an affected family member.
Inheritance is autosomal dominant (one only need to get the abnormal gene from one parent in order to inherit the disease). Genetic counseling should be offered.
Treatment requires a coordinated multidisciplinary approach. Surgery is the mainstay of treatment for tumors. Management should include lifelong surveillance (ophthalmologic, MRI brain and abdominal scans, laboratory testing). At-risk relatives should be entered into a screening program in childhood unless VHL is excluded by molecular genetic testing.
Prognosis depends on the occurrence of multiple tumors. RCC is the main cause of death, followed by CNS hemangioblastomas. Average life expectancy is estimated to be 50 years; however, regular surveillance and early detection and management of tumors reduce morbidity and mortality.
Source: Pr Eamonn MAHER. Orphanet