“Our findings have the potential to greatly widen treatment options for the vast majority of severe aplastic anemia patients,” according to Robert Brodsky, M.D., professor of medicine and oncology at the Johns Hopkins Kimmel Cancer Center and an author of the report.
Results of the small clinical trial have already prompted the organization of a larger national trial being led by Amy DeZern, M.D., an assistant professor of oncology and medicine at the Johns Hopkins Kimmel Cancer Center, with plans to involve patients at 25 medical centers across the country.
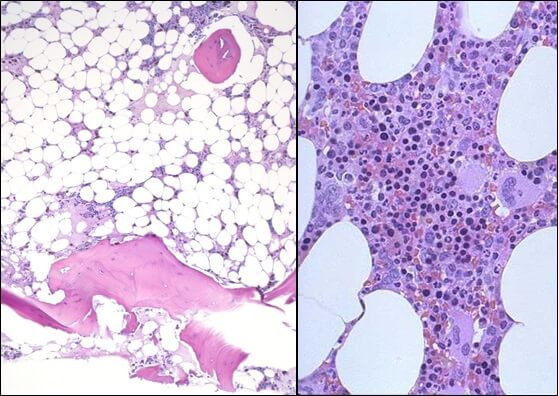
Diagnosed in about one in 250,000 people each year, aplastic anemia occurs when one’s own immune system damages blood-making bone marrow cells, which gradually stop producing red and white blood cells and platelets.
Patients must receive frequent blood transfusions, take multiple medicines to suppress the autoimmune response that damages the marrow, take other drugs to prevent infections, and limit contact with the outside world to avoid infection and even minor injury. Over the long term, most patients eventually die of infections.
When immunosuppressive therapy fails to keep the disease in check — in as many as 30 to 40 percent of patients — doctors usually prescribe a drug called eltrombopag, which is used in a variety of blood disorders to increase platelets. The drug, according to the Johns Hopkins experts, works only in about 30 percent of patients and usually leads to a partial, not complete, response.
Brodsky and DeZern say that the only curative treatment is a bone marrow transplant, but few patients have donors who are “fully matched” — sharing the same collection of immune-stimulating proteins that decorate every cell in the body.
In an effort to overcome the donor shortage and offer transplant to more patients, DeZern, Brodsky and their colleagues enrolled 16 patients between 11 and 69 years of age in this study from July 2011 through August 2016.
Each of the patients had failed to respond to immunosuppressive therapy or other drug treatments. None had access to a related fully matched bone marrow donor but did have an available and willing donor who was a half match. Three patients used unrelated donors.
After administering a cocktail of drugs designed to suppress their immune system and prevent rejection of the donor marrow, the patients received half-matched bone marrow transplants, some from siblings or parents, and others from unrelated donors.
Three and four days after their transplants, the patients received high doses of the chemotherapy drug cyclophosphamide. For the next year, or slightly longer, they remained on immunosuppressive medications, including tacrolimus, then stopped taking them.
Within weeks of their transplants, tests showed that each of the patients’ red and white blood cell and platelet counts had returned to normal levels without the need for blood transfusions. Once immunosuppressive therapy was stopped, none of the patients required further treatment related to their disease, the Johns Hopkins team reported.
Although 13 patients were able to discontinue immunosuppressive drugs a year after their transplant, three developed mild graft-versus-host disease (GVHD), a common complication of bone marrow transplants that occurs when immune cells in the transplant attack the newly transplanted cells. Two patients had mild GVHD that appeared on their skin, and one patient’s GVHD occurred in the mouth and skin. After a few extra months of immunosuppressive therapy, their GVHD subsided, and they also were able to stop taking these medications.
Ending all therapy related to their disease has been life-changing for the patients, says DeZern. “It’s like night and day,” she says. “They go from not knowing if they have a future to hoping for what they’d hoped for before they got sick. It’s that transformative.”
Successful transplants using partial match donors, Brodsky says, open up the transplant option to nearly all patients with this condition, especially minority patients. Seven of the 16 patients treated at Johns Hopkins self-identified as nonwhite.
A full sibling only has a 25 percent chance of being a full match. However, 100 percent of parents and 50 percent of siblings or half-siblings are half matches, regardless of ethnicity. The average person in the United States has about four half matches or better. “Now, a therapy that used to be available to 25 to 30 percent of patients with severe aplastic anemia is potentially available to more than 95 percent,” says Brodsky.
The idea of using cyclophosphamide after a partial-match transplant was first pioneered decades ago by Johns Hopkins Kimmel Cancer Center experts. Brodsky says the drug destroys patient’s diseased immune system cells but does not harm the donor’s blood stem cells, which create new disease-free blood cells in the patient.
Bone marrow transplants are costly — sometimes exceeding more than $300,000. However, Brodsky and DeZern say that full and half-matched transplants are life-saving for many, and there is cost-saving potential when aplastic anemia patients can avoid a lifetime of immunosuppressive therapy, hospitalizations, medications and blood transfusions.
Other Johns Hopkins researchers who participated in the study include Marianna Zahurak, Heather Symons, Kenneth Cooke and Richard J. Jones.
This research was supported by the National Institutes of Health (K23 HL123601-01).