Researchers at Johns Hopkins Medicine have transformed skin cells from patients with Lou Gehrig’s disease, or amyotrophic lateral sclerosis (ALS), into brain cells affected by the progressive, fatal disease and deposited those human-made cells into the first public ALS cell library, enabling scientists to better study the disease. Using a genetic engineering technique that causes adult skin cells to transform into “pluripotent” cells, otherwise known as induced pluripotent stem cells, which can take the form of many different cells found in other parts of the body, “we make brain cells out of the patient’s own skin,” says Jeffrey Rothstein, M.D., Ph.D., who directs the Brain Science Institute and the Robert Packard Center for ALS Research.
While the technique for creating these human-made cells has been used by other researchers, Rothstein and his colleagues are the first to use these induced pluripotent stem cells to create the largest library of brain cell lines donated voluntarily by more than 20 ALS patients whose disease was caused by various genetic mutations. “These human cellular tools will serve as a platform to understand ALS and someday discover new drugs to treat our patients,” says Rothstein, senior author of a study about the work, which was recently published online in PLOS ONE.
More than 30,000 people in the U.S. are diagnosed with ALS. Men appear to be affected slightly more than women. One of every 500 deaths in men is due to ALS, says Rothstein, “so just about everyone is going to know a neighbor, friend or family member who will eventually succumb to this terrible disease.”
There is no known cure for ALS and only one FDA-approved drug, riluzole, which may only add a year to a patient’s life span, says Rothstein.
Since the 1990s, researchers have studied the disease and its potential treatments in mice. The mouse model looks very much like what happens in people, Rothstein says, “but after 25 years, it has not led to the development of a drug that works in our patients.” A handful of drugs were effective in mice and passed phase II clinical trials, which establish a drug’s safe dosage for humans, but they all failed phase III trials, which confirm a drug’s effectiveness in people.
“There has to be a sea change in how we approach ALS,” Rothstein says.
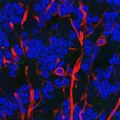
Because there is no ethical or simple way to obtain brain tissue from living ALS patients, Rothstein and colleagues turned to induced pluripotent stem cell production. The technique gives researchers a tool to look at diseased human brain cells, including specialized nerve cells called astroglia, which play a critical role in ALS progression. Rothstein and his team created 22 patient-specific cell lines that included some common mutations known to be associated with ALS and deposited them in a cell library, to be shared with other scientists.
The library includes cells from patients with inherited ALS, which accounts for about 10 percent of ALS cases. Rothstein and his team also have generated cells from patients with the noninherited form of the disease — sporadic ALS, which makes up 90 to 95 percent of ALS cases. From one patient, the researchers collected a genetic variant found in both inherited and sporadic forms of the disease, and they added that variant to the library.
Many scientists around the world have already used the library, and Rothstein hopes it will grow, with researchers making deposits of their own patients’ cell lines. Eventually, induced pluripotent stem cells may be used to model diseases other than ALS and to test potential drug treatments, says Rothstein, who adds: “Now we have a real model for what’s wrong with my patients.”