Source: Nadia BELMATOUG MD; Jérôme STIRNEMANN
MD.
Orphanet
Gaucher disease is a rare inherited disorder, in which people do not
have enough of an enzyme called acid beta-glucosidase, which normally
breaks down a fatty waste product called glucosylceramide. Without the
enzyme, glucosylceramide builds up in the body, typically in the liver,
spleen and bone marrow, which produces the symptoms of the disease:
anaemia (low red blood cell counts), tiredness, easy bruising and a
tendency to bleed, an enlarged spleen and liver, and bone pain and
breaks.
Gaucher disease (GD) is a lysosomal storage disorder encompassing three
main forms (types 1, 2 and 3), a fetal form and a variant with cardiac
involvement (Gaucher disease - ophthalmoplegia - cardiovascular
calcification or Gaucher-like disease).
Frequency
The frequency is approximately 1/100,000. The annual incidence of GD in the
general population is about 1/60,000, but it can reach up to 1/1,000 in
Ashkenazi Jewish populations.
Symptoms
The clinical manifestations of
this disease are highly variable.
- Type 1 (90% of cases) is the
chronic and non-neurological form associated with organomegaly (spleen,
liver), bone anomalies (pain, osteonecrosis, pathological fractures) and
cytopenia.
- Type 2, the acute neurological form, is characterized by
early onset, rapidly progressing brainstem dysfunction, associated with
organomegaly and leading to death before the age of 2.
- Type 3, the
subacute neurological form, affects children or adolescents and is
characterized by progressive encephalopathy (oculomotor apraxia,
epilepsy and ataxia) with the systemic manifestations seen in type 1.
The fetal form manifests with a decrease or absence of fetal movements
or anasarca. Gaucher-like disease presents with progressive
calcification of the aorta and the aortic and/ or mitral valves as its
main feature.
Genetics
GD is due to mutations in the
GBA gene (1q21) that codes for a lysosomal enzyme, glucocerebrosidase, or in very rare cases the
PSAP
gene that codes for its activator protein (saposin C). The deficiency
in glucocerebrosidase leads to the accumulation of glucosylceramidase
(or beta-glucocerebrosidase) deposits in the cells of the
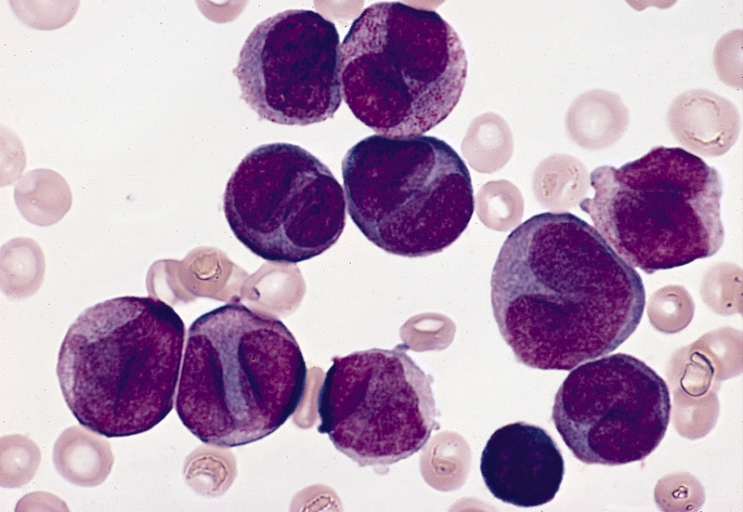
reticuloendothelial system of the liver, the spleen and the bone marrow
(Gaucher cells).
Transmission is autosomal recessive.
Diagnosis
Formal diagnosis of the disease is determined by
the measurement of glucocerebrosidase levels in circulating leukocytes.
Genotyping confirms the diagnosis.
Differential diagnoses
include other lysosomal storage disorders. The presence of Gaucher-like
cells can be found in certain hematologic diseases (lymphoma, Hodgkin's
lymphoma and chronic lymphocytic leukemia; see these terms).
Treatments
There
are two available treatments for GD type 1 and 3: enzyme substitution
therapy (using
imiglucerase or
velaglucerase) and substrate reduction
therapy (
miglustat). These treatments are ineffective for GD type 2.
Prognosis
The
prognosis is good in GD type 1.
In type 2, death usually occurs before
the age of 2. Without specific treatment,
GD type 3 progresses to death
within a few years.


.jpg)