
Our bodies contain thousands of active substances called enzymes. For example, your stomach has enzymes that help break down the food you eat. Similarly, the enzyme acid alpha-glucosidase (pronounced AL-fa glue-CO-sigh-days), also called GAA, breaks down glycogen, a form of sugar that is stored in a specialized compartment of muscle cells throughout the body. People with Pompe do not have enough GAA enzyme, so glycogen builds up in muscle cells. This leads to progressive loss of muscle function. Pompe mostly affects the skeletal (including heart) and respiratory muscles.
There are two types of Pompe: infantile-onset and late-onset. Infantile-onset is the result of complete or near complete deficiency of GAA. Late-onset is caused by partial deficiency of GAA.
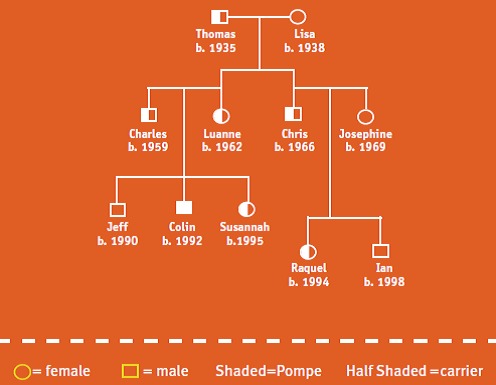
How is Pompe passed on?
Pompe is an autosomal recessive condition. This means two non-working copies of the gene are needed for an individual to be affected by the condition. Individuals with one working copy and one non-working copy of the gene are called carriers.
• When both parents are carriers, there is a 1 in 4 (25%) chance that any child born will have Pompe.
• If one parent has the disease and the other parent is a carrier, there is a 1 in 2 (50%) chance that any child born will have Pompe.
• When both parents have Pompe, all of their children will inherit the disease.
Colin’s story

My name is Colin, and I have Pompe. No one else in my family has Pompe.
My grandfather, Thomas, was a carrier. He passed the gene to my mom and uncle Chris; they are both carriers, too. My uncle married Josephine, who does not have the gene change for Pompe. They have two children – my cousin Raquel is a carrier, and Ian is not.
My mom married another carrier—my dad. This means that each of their children—me, my brother and sister—had a 1 in 4 (25%) chance of having Pompe, a 1 in 2 (50%) chance of being a carrier, and a 1 in 4 (25%) chance of not having the Pompe gene at all. Jeff was born first—he doesn’t have the Pompe gene change! I’m the middle child—I have Pompe. Susannah is a carrier.
Infantile-onset
Infants affected by Pompe show symptoms soon after birth, and the disease progresses rapidly. Most babies die from cardiac or respiratory (breathing or lung-related) complications before their first birthday.
Major signs and symptoms
- Muscle weakness or a “floppy” appearance
- Poor muscle tone
- Enlarged heart (usually detected by X-ray)
- Breathing problems and lung infections
- Feeding problems and failure to grow at the expected rate (failure to thrive)
Kerri’s story

My daughter Sarah was a short-lived, sweet, beautiful child. Four months after she was born in 1996 we found out she had Pompe. Five months after that she passed away. It’s a genetic disease, and she had my genes, so I blamed myself. More than a decade later, I was having health problems. I was misdiagnosed with chronic fatigue syndrome, arthritis, depression, and more.
I told my doctor I could not go on living like this. He referred me to a neurologist, and I finally got a diagnosis that stuck: Pompe.

I was upset when I found out. I didn’t want it to be Pompe. I was terrified to tell my family and friends, but it was comforting that I could reassure them that this was not the death sentence Sarah was given. I could not let myself have a pity party. I prayed a lot and did a lot of investigating. Someone told me early on that I would be my own best advocate. That was the best advice I was given.
Late-onset
Though late-onset Pompe may appear more gradually than the infantile-onset form, it is always progressive (gets worse over time) and is often debilitating. The onset can be as early as the second year of life or as late as the sixth decade of adulthood. In general, the rate of disease progression is variable in the late-onset form, but a rapid decline can occur at any time and is hard to predict. The primary symptom is muscle weakness, which may include respiratory weakness and failure. The heart is usually not involved.
Major signs and symptoms
- Weakness in the limbs and trunk
- Weakness of the diaphragm and other respiratory muscles
- Weakness in the hip muscles, which can lead to difficulty walking and climbing stairs
- Fatigue (tiredness)
- Reduction in quality of life, such as the ability to work, perform household chores, and move independently outside the home
Testing and Diagnosis
Accurate diagnosis and treatment are important for people with Pompe because the disease is always progressive. Because the severity of signs and symptoms, rate of disease progression, and age of symptom onset can vary widely among people with Pompe, it is often mistaken for other disorders with similar signs and symptoms such as:• Dystrophies (e.g., limb-girdle muscular dystrophy)
• Congenital myopathies
• Motor neuron disorders (neurological disorders that destroy motor neurons, the cells that control voluntary muscle movement such as speaking, walking, breathing, and swallowing; e.g., spinal muscular atrophy)
Doctors may use a variety of tests to assess your condition, such as muscle and movement tests, breathing tests, and laboratory tests. All of these tests reveal some aspect of Pompe, but none of these tests alone can specifically identify the condition. Once your doctor suspects Pompe, he or she can screen for Pompe with a simple blood test that measures GAA enzyme activity in the blood. All people with Pompe have lower than normal GAA activity levels.
Another testing option, called genotyping, uses DNA from a blood sample to see if the person has the gene mutation, or defect, that causes Pompe disease. This is especially useful within families in which the particular mutation is known, or to identify carriers of the disease.
Once Pompe disease is diagnosed, testing of all family members and a consultation with a genetics professional are recommended.
Source: Genetic Alliance